Abstract
Hepatocellular carcinoma (HCC) is an aggressive malignancy with increasing morbidity and mortality worldwide. The migration and motility of HCC tumor cells are enhanced by the formation of invadopodia, which comprise membrane protrusions at the leading edge. Previous studies have showed that cell division cycle 42 (CDC42) plays an essential role in remodeling the cytoskeleton, which is associated with invadopodia formation and thus mediates cellular movement. Therefore, aberrant expression of CDC42 is hypothesized to promote tumor cell migration. In this review, we discuss the important role of CDC42 activation induced by guanine nucleotide-exchange factors (GEFs), which function as upstream regulators to activate CDC42, thereby mediating HCC invasion and metastasis by facilitating invadopodia formation. Furthermore, inhibitors targeting the CDC42-GEF interaction might be developed as an intervention against HCC metastasis.
Keywords
CDC42, GEFs, HCC, Cell motility, Invadopodia, Metastasis, Inhibitors
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common cancer worldwide, as well as the third leading cause of cancer-related deaths [1], with aggressive malignancy and high invasiveness. The extravasation of HCC cells plays an important role in distant metastasis; however, its precise mechanism remains unclear. Invadopodia, which are associated with invasive tumor cells, are crucial for cellular invasion and metastasis [2]. During the process of HCC invasion and metastasis, invadopodia formation occurs in tumor cells, thus enhancing the cellular motility.
Cell division cycle 42 (CDC42) is a 21.3 kDa Rho GTPase protein, which is activated when bound to GTP and inactivated when bound to GDP [3]. The release of GDP requires the participation of a guanine nucleotide-exchange factor (GEF). GEFs bind to CDC42-GDP and induce the exchange of GDP for GTP to activate CDC42 [4]. Subsequently, active CDC42-GTP interacts with its specific effectors and triggers biological effects, including cell migration. CDC42 is aberrantly activated in many human cancers, and multiple biological functions mediated by CDC42 are associated with tumor metastasis [5].
In this review, we summarize the role of active CDC42 induced by GEFs in HCC metastasis. Certain inhibitors targeting the Rho GEFs that are involved in aberrant CDC42 activation (thus mediating HCC invasion and metastasis), which might provide new insights into the prevention and treatment of HCC metastasis.
CDC42 Plays an Essential Role in Cell Motility
The Rho GTPases form a subfamily of the RAS superfamily of small GTP-binding proteins with a size of 21 to 25 kDa [6]. These proteins function as molecular switches that trigger signal transduction cascades. Rho GTPases have recently been expanded to over 20 members [7], Among them, CDC42 has been extensively studied.
CDC42 is a key signal transduction protein that plays a central role in intracellular signaling networks, mediating a variety of signal transduction pathways initiated by growth factors, cytokines, and cell adhesion proteins [8]. CDC42 is mainly regulated by four classes of regulatory proteins: guanine nucleotide exchange factor (GEF), GTPase-accelerating protein (GAP), regulator of G protein-signaling (RGS), and guanine nucleotide dissociation inhibitor (GDI) (Figure 1). As a molecular switch, CDC42 shows an active "on" form when bound to GTP and an inactive "off" form when combined with GDP. The conversion of the inactive GDP-bound form to the active GTP-bound form is mediated by GEFs [9]. In addition, the hydrolysis rate of GTP is promoted by GAP and RGS, but inhibited by GDI. In response to external signals, GEFs engage CDC42 and form macromolecular complexes with scaffolding proteins and/or kinases, resulting in CDC42 activation [3]. Activated CDC42 then mediates a signaling cascade leading to the activation of various downstream effectors that influence many cellular processes, such as cell adhesion, motility, polarity, cytokinesis, and growth [7-12].
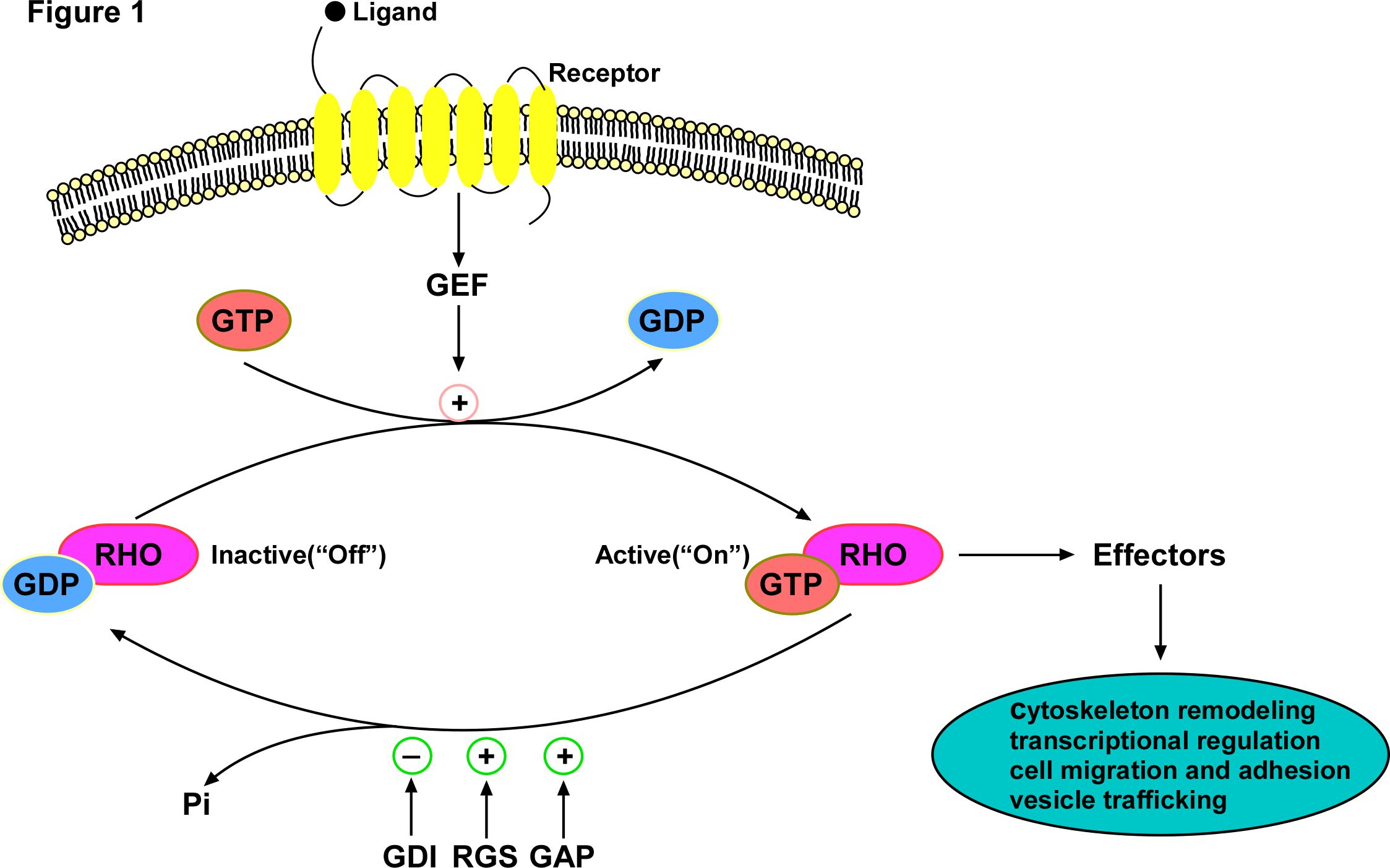
Figure 1. The classical RHO GTPase cycle: Rho GTPases typically cycle between the active GTP-bound form and the inactive GDP-bound form. This GTP-binding/GTP-hydrolysis cycle is primarily regulated by four regulators: GEF, GAP, RGS and GDI, which GEF activates the Rho GTPase via catalyzing the exchange of GDP to GTP, while the GAP and RGS inactivate the Rho GTPase by hydrolyzing the GTP, thereby acting as inhibitors of Rho GTPases. The GDIs sequester and extract the Rho GTPases from the membrane to prevent the interactions between Rho and GEFs, GAPs, RGS and downstream effectors. Activated Rho GTPase turns on effectors to transduce signals leading to signal transduction cascades and triggers biological effects such as cytoskeleton remodeling, polarity, migration and adhesion. With the hydrolysis of bound GTP to form GDP and Pi, Rho GTPase returns to the inactive form.
Cell motility is a multistep process that includes a migration process involving the formation of protrusions at the leading edge [13]. The initial step of cell migration is extension of the cytoplasm, which is induced by actin polymerization at the leading edge. Finger-like structures called filopodia (the actin-based structure of membrane protrusions at the cell periphery) are induced by CDC42 [14,15]. During the process of membrane protrusion, new actin polymerization is involved and actin nucleators, such as Wiskott–Aldrich syndrome protein (WASP), neuronal-WASP (N-WASP) and the actin-related protein-2/3 (Arp2/3) complex, are required [16-18]. WASP family proteins function as scaffold proteins that link upstream signals to activate the Arp2/3 complex, the initiation complex for microfilament assembly, which leads to a burst of actin polymerization. The first identified member of the Wiskott–Aldrich syndrome protein (WASP) family was WASP itself, which was initially found as the causative gene of WAS, an X-linked recessive disease that was described as a clinical trial of immunodeficiency, thrombocytopenia, and eczema [19]; and N-WASP, the second member of the WASP subfamily, was identified in the brain but widely expressed in different tissues, which shares several functional motifs with WASP. Under resting conditions, WASP and N-WASP are inactive forms due to the interaction between the GTPase-binding domain and the carboxy-terminal region, thereby inhibiting the binding of the Arp2/3 complex [20]. Once activated by various external stimuli, CDC42-GTP activates WASP and N-WASP via relief of the autoinhibitory mechanism. Thus, CDC42 contributes to the indirect activation of the Arp2/3 complex to initiate peripheral actin polymerization, thereby leading to filopodia formation [21] (Figure 2).
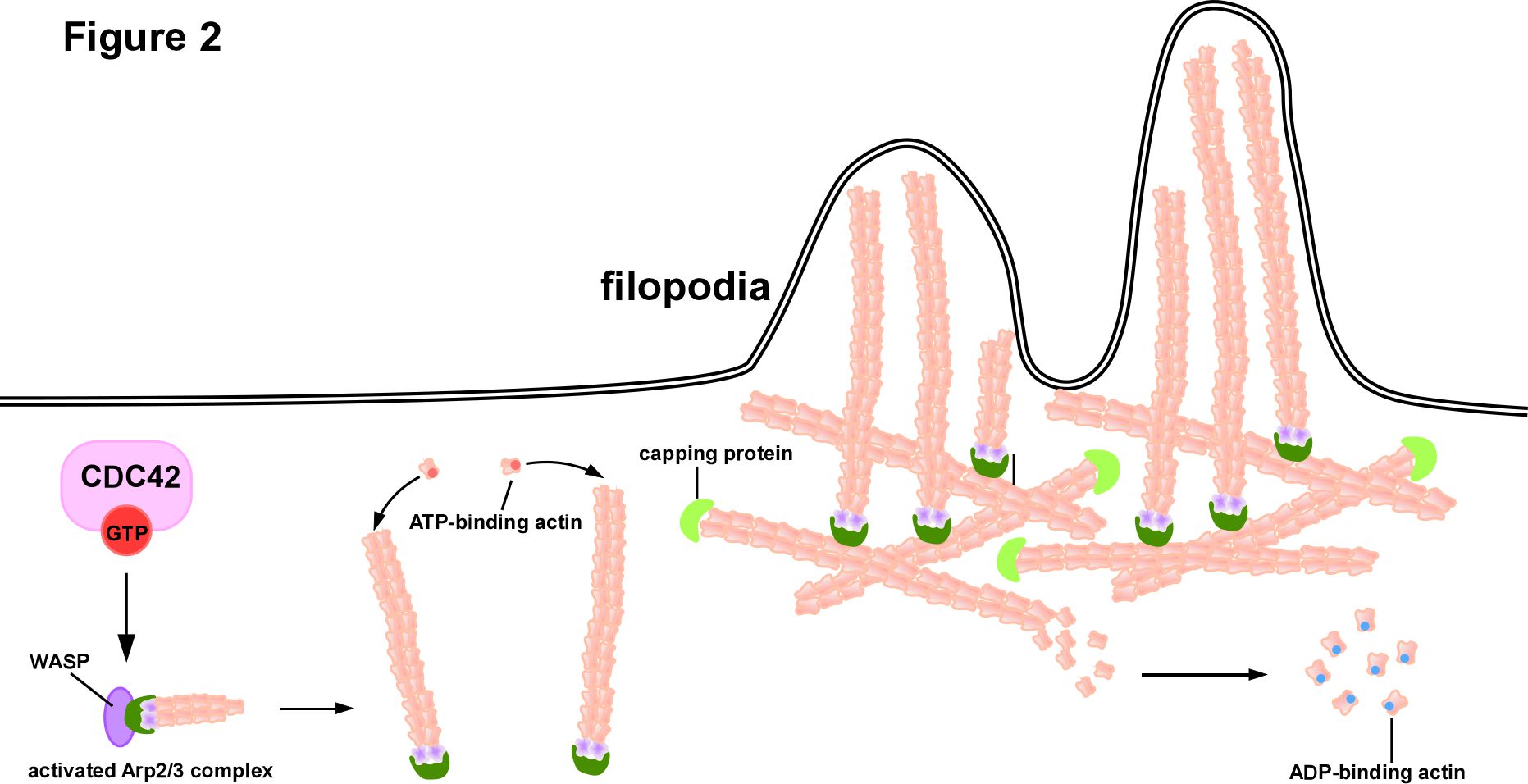
Figure 2. CDC42-driven filopodia formation mode: Extracellular signals bind to receptors located on the cell membrane to initiate intracellular signals and activate CDC42. Activated CDC42 activates Wiskott–Aldrich syndrome protein (WASP) via relief of the autoinhibitory mechanism, then Arp2/3 complex is activated thus initiates the assemble of microfilaments. Only ATP-binding actin can participate in microfilament assembly. Once bound to the microfilament ends, actin exerts its ATPase activity to hydrolyze ATP to ADP and releases the ADP-binding actin. Capping proteins bind to the ends of microfilaments to prevent their depolymerization or over assembly. The continuous extension of microfilaments in length pushes the cytoplasmic membrane to form filopodia.
Furthermore, a variety of other molecules are required to direct the CDC42-dependent formation of filopodia. The interaction of CDC42 with IRSp53 (an SH3 domain-containing scaffold protein) allows the recruitment of Mena to the IRSp53 SH3 domain, which initiates actin filament assembly into filopodia [22]. In addition, CDC42 phosphorylates and activates LIM motif-containing protein kinase (LIMK) via P21 activated kinase 1 (PAK1), which in turn efficiently induces the phosphorylation and inactivation of cofilin, resulting in decreased depolymerization of F-actin, ultimately leading to filopodia formation [23]. Many effectors of CDC42 also function downstream of RAC [14]; consequently, CDC42 is considered a potential regulator that drives the activity of RAC-dependent lamellipodia, suggesting a molecular model for the coordinated control of cell motility. In conclusion, the basic mechanism of CDC42 in cell motility is that CDC42 is activated by GEFs and then induces the formation of a protrusion at the leading edge to facilitate cell migration.
CDC42 in HCC Invasion and Metastasis
HCC is a common cause of cancer?related mortality and morbidity worldwide. Despite the substantial improvement in novel diagnostic methods and therapeutic strategies, the prognosis of patients with HCC is still unsatisfactory [1], with median 5 years survival rate of only 18% [24]. Moreover, for patients with advanced HCC, the median survival is only 6-8 months [25], and the agent that has been shown to improve survival, sorafenib, can only extend the median overall survival to 10.7-14.7 months [26]. The poor prognosis of HCC is due to its aggressive invasiveness and high metastasis. The lung is the most common site of extrahepatic metastasis of HCC [27]. In the past, it was considered that the pulmonary metastases of HCC were mostly multiple lesions, which did not have surgical indications, and comprehensive non-surgical treatment was often adopted [28]. However, non-surgical therapies including systemic chemotherapy for pulmonary metastasis of HCC have limited efficacy. Furthermore, tumor cell extravasation is a key step during cancer metastasis, yet the precise mechanisms are unclear, and there is no consensus on the treatment strategy for pulmonary metastasis of HCC. Therefore, there is an urgent need to determine the underlying mechanism of extravasation in HCC pulmonary metastasis.
Cell motility changes markedly during tumor cell invasion and metastasis, and is always deregulated in metastatic cancer. Given that RHO GTPases participate in various cellular functions, it is believed that they are connected with almost every stage of cancer development and progression, including tissue invasion and metastasis [29]. Previous studies have demonstrated frequent dysregulation of CDC42 in numerous human cancers, which is mainly caused by dysregulation of its upstream regulators, especially GEFs [30]. CDC42 is presumed to facilitate the aggressive biological behaviors of tumor cells via remodeling the cytoskeleton, which affects cell motility. Recent studies indicated that CDC42 participates in the tumorigenesis and progression of HCC [6]. Here, we discuss the molecular mechanism by which CDC42 plays an essential role in migration and metastasis by affecting the cell motility.
CDC42-induced modulation of cell motility is involved in the underlying molecular mechanism of HCC invasion and metastasis. Strikingly, CDC42 was found to enhance the ability of HCC cells to invade surrounding tissues by inducing filopodia formation [31]. In our latest published study, we found that the upregulated expression of Rho guanine nucleotide exchange factor 37 (ARHGEF37) correlated positively with high risk of HCC pulmonary metastasis [32]. Furthermore, we demonstrated that overexpression of ARHGEF37 increased the CDC42-GTP level in HCC cells, and enhanced their extravasation and lung metastatic capability by promoting the formation of invadopodium, a special F-actin-rich matrix-degrading structure, arises on the ventral surface of the cell membrane and found in a variety of carcinomatous cells whereas filopodia appears on the leading edges of migrating cells and function to command the direction of migration [33], consequently disrupting the interaction between endothelial cells and pericytes [32]. Meanwhile, overexpression of the GEFs faciogenital dysplasia 1 (FGD1) in HCC leads to CDC42 activation, thus facilitating invadopodia formation and extracellular matrix (ECM) degradation, and subsequent tumor cell invasion [34]. Moreover, RICH2, (Rho GTPase activating protein 44, also known as ARHGAP44), regulates the formation of filopodia in HCC in a CDC42 dependent manner, and stable overexpression of RICH2 significantly inhibited the invasion of HCC cells in vitro [35]. In addition to upstream regulators of CDC42, other cellular components are also involved in HCC invasion and metastasis by activating the CDC42 axis. For example, CDC42 interacting protein 4 (CIP4) is a CDC42 effector that assists membrane deformation and actin polymerization. Specifically, phosphorylation at T225 on CIP4 facilitates the interaction with CDC42, thus promoting the formation of functional invadopodia, which is involved in regulating cancer cell invasiveness and metastasis [36].
In brief, the molecular mechanism by which CDC42 mediates HCC invasion and metastasis is mainly regulated by the overexpression of upstream regulators, especially GEFs, thus facilitating invadopodia formation, ECM degradation, and subsequent tumor cell invasion.
Treatment Strategies Targeting the CDC42-GEF Interaction
Until recently, Rho GTPases themselves were deemed to be “undruggable” considering their inaccessible structure and multifaceted function [37]. However, the upstream regulators of Rho GTPases play a crucial role in tumor invasion and metastasis, thus making them potential targets to develop anti-tumor agents. Although no therapeutically effective drugs targeting CDC42 signaling for cancer treatment are available, small molecule inhibitors targeting the CDC42 axis combined with other anti?cancer agents could be promising and of potent according to recent studies [30,38-42].
Unlike protein kinases, which can be targeted by their nucleotide analogs, it is difficult to target Rho GTPases by such small molecule modulations because of the micromolar GTP concentration in cells and the sub-nanomolar binding affinity of Rho GTPases for GTP or GDP [37]. Given that Rho GTPases are activated by different RhoGEFs, which respond to different signals in a temporally and spatially manner, targeting specific RhoGEFs provides feasible selectivity. Several small molecule inhibitors targeting specific RhoGEFs and preventing their binding to Rho GTPases have been developed. For example, compound CID2950007 and its analog CID44216842 were synthesized, both of which inhibited GTP binding to CDC42 in a dose?dependent manner [40]. Inhibition of CDC42 activation induced by CID2950007 effectively inhibited filopodia formation and led to ineffective cell migration in human ovarian cancer cells. In addition, intersectin 1 (ITSN1) has been identified as a CDC42-specific GEF using in vitro studies [43] and the small molecule inhibitor ZCL278 can dock into the ITSN1 binding groove of CDC42 [39]. ZCL278, synthesized and purified based on the CDC42-ITSN1 interaction interface, is able to fit into the surface groove of CDC42, which is critical for GEF binding, with low micromolar affinity (Kd = 6.4 μM) [39]. And it suppresses CDC42-mediated actin-based motility [32] and migration [44] without disrupting cell viability, thus providing a powerful tool for researching the interaction of CDC42-GEF in human pathogenesis, such as those of cancer. In our latest published study, we demonstrated that ARHGEF37, the most salient molecules screened through integrated two public datasets of pulmonary metastasis in HCC, The Cancer Genome Atlas (TCGA) HCC dataset and online public Kaplan-Meier Plotter database, while ITSN1 was not in the candidate list, catalyzed the release of GDP from CDC42 to bind with GTP, activating CDC42 to induce the formation of invadopodium at the leading edge, which greatly enhanced cell migration, leading to pulmonary metastasis of HCC. Importantly, treatment with ZCL278 dramatically inhibited that active CDC42 induced by ARHGEF37 mediated HCC invasion and metastasis (Figure 3). Our findings further confirmed that the inhibition of the CDC42?GEF interaction by the small molecule ZCL278 impedes HCC invasion and metastasis. Taken together, inhibitors targeting the CDC42-GEF interaction to inhibit tumor invasion and metastasis will be an excellent candidate for lead compound optimization in further anticancer studies.
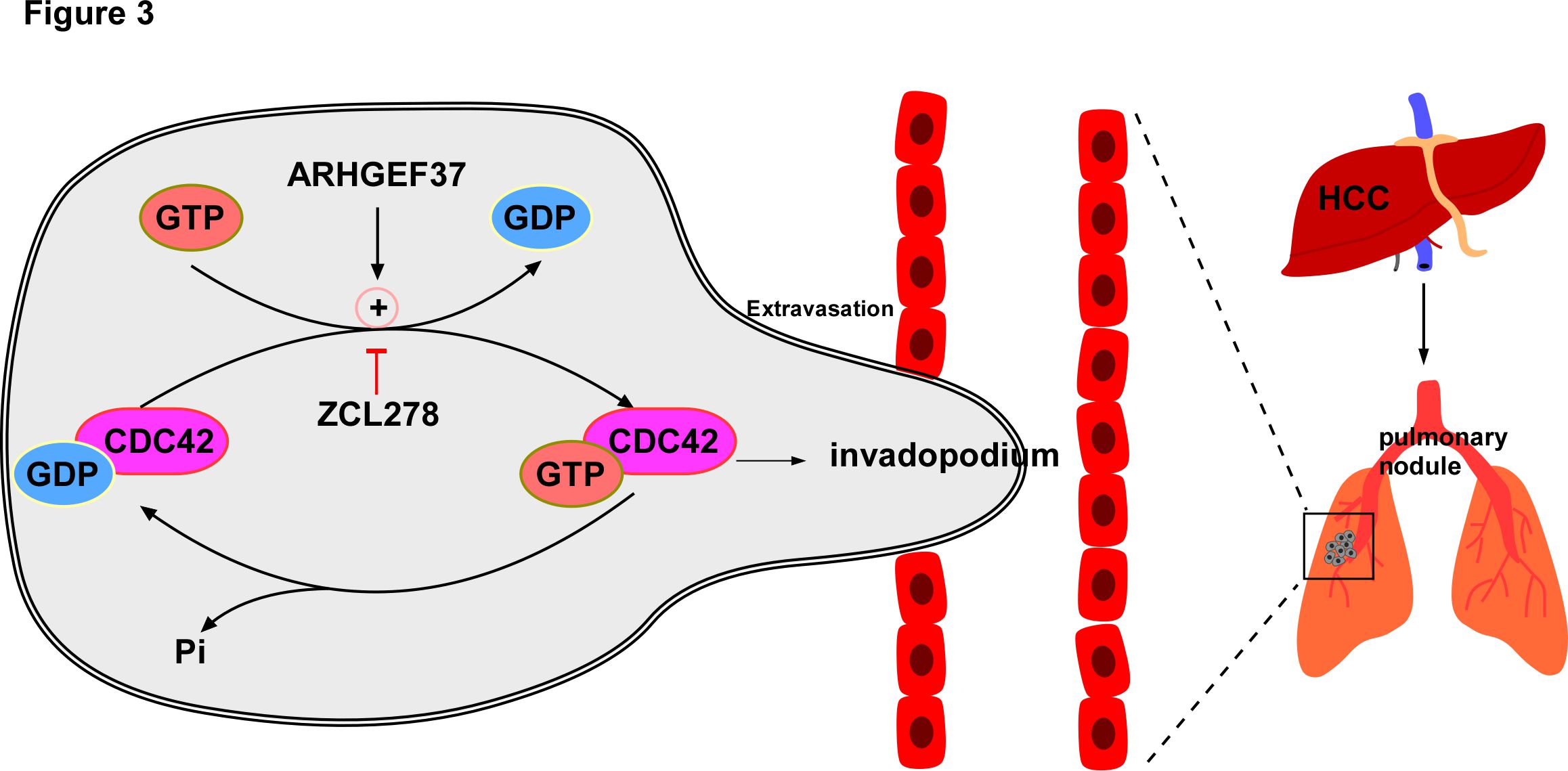
Figure 3. Excessive activation of CDC42 caused by ARHGEF37 overexpression promotes HCC extraversion and pulmonary metastasis via invadopodia formation. ARHGEF37 activates CDC42 via catalyzing CDC42-GDP from to CDC42-GTP from, resulting in a conformational change. With the hydrolysis of bound GTP to form GDP and Pi, CDC42 returns to the inactive form. Activated CDC42 induces the formation of invadopodium at the leading edge, which greatly enhances cell migratory capability, leading to extravasation and promoting pulmonary metastasis of HCC. ZCL278, the specific inhibitor targeting CDC42-GEF interaction, impedes HCC invasion and metastasis.
Conclusion
The Rho family GTPase CDC42 regulates cytoskeletal organization in physiological processes, including cell proliferation, motility, and polarity. The activation of CDC42 in response to upstream signals is mediated by GEFs, which convert the GDP-bound inactive form to the GTP-bound active form of CDC42. Activated CDC42 transduces signals to downstream effectors, the autoinhibitory mechanism of WASP/N-WASP was relieved and thus effectual, then Arp2/3 complex is activated thus initiates the assemble of microfilaments, the continuous extension of microfilaments in length pushes the cytoplasmic membrane to form invadopodia; and generates cellular effects, such as cytoskeleton remodeling, polarity, migration and adhesion. Aberrant activation of CDC42 results in pathogenesis, such as tumor invasion and metastasis. Tumor invasion requires orchestration of actin-based protrusions capable of ECM degradation and cell locomotion. In numerous human tumors, including HCC, the ability of cells to form invadopodia and their invasive potential are directly correlated.
Recent studies indicate that CDC42 plays an essential role in hepatocellular carcinoma invasion and metastasis [6]. Mechanistically, CDC42-mediated HCC invasion and metastasis are mainly regulated by the overexpression of upstream regulators, particularly GEFs, which thus facilitates invadopodia formation, ECM degradation and subsequent tumor cell invasion. Based on the molecular mechanism, novel strategies targeting CDC42-GEF interactions might lead to drugs to prevent and treat HCC invasion and metastasis. Of particular interest, in our latest published study, we identified ARHGEF37, a specific CDC42 guanine nucleotide exchange factor, catalyzes the release of GDP from CDC42 to bind with GTP, which activates CDC42 by triggering a conformational change, resulting in the formation of invadopodia, which greatly enhances cell migration, leading to extravasation and promoting pulmonary metastasis of HCC. ZCL278, the specific inhibitor targeting CDC42-GEF interaction, impedes HCC invasion and metastasis [32]. Therefore, our study provides a new insight into the underlying mechanisms on the ARHGEF37 overexpression-mediated extravasation and pulmonary metastasis of HCC, indicating that agents targeting CDC42-GEF interaction should be of potency in therapeutic interventions of HCC metastasis.
Several inhibitors that interfere with CDC42's role on cellular motility have been identified, such as compound CID2950007 and its analog CID44216842, and ZCL278, although none have been applied to clinical therapeutics. The regulatory mechanism of the RHO GTPase family should be further clarified to permit the design of additional therapeutic interventions and more attention should be paid to the design of inhibitors targeting RHO GTPase effectors and regulators. Moreover, further studies are needed to understand the detailed roles of RHO GTPases in the development of HCC.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This work was supported by Natural Science Foundation of China (No. 82030078, 81830082, 82203746); Natural Science Foundation of Guangdong Province (No. 2023A1515010370); the China Postdoctoral Science Foundation Grant (No. 2022M723644).
Author Contributions Statement
M.T. and R.F. wrote the manuscript text. R.F. created the figure and edited the manuscript for important intellectual content. M.T. and J.L. supervised the study. All authors read and approved the final manuscript.
References
2. Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 2014;8(5):1558-70.
3. Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496-510.
4. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6(2):167-80.
5. Arias-Romero LE, Chernoff J. Targeting Cdc42 in cancer. Expert Opin Ther Targets. 2013;17(11):1263-73.
6. Wang T, Rao D, Yu C, Sheng J, Luo Y, Xia L, et al. RHO GTPase family in hepatocellular carcinoma. Exp Hematol Oncol. 2022;11(1):91.
7. Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093-101.
8. Cerione RA. Cdc42: new roads to travel. Trends Cell Biol. 2004;14(3):127-32.
9. Cerione RA, Zheng Y. The Dbl family of oncogenes. Curr Opin Cell Biol. 1996;8(2):216-22.
10. Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509-14.
11. Etienne-Manneville S. Cdc42--the centre of polarity. J Cell Sci. 2004;117(Pt 8):1291-300.
12. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690-701.
13. Machesky LM, Li A. Fascin: Invasive filopodia promoting metastasis. Commun Integr Biol. 2010;3(3):263-70.
14. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81(1):53-62.
15. Gupton SL, Gertler FB. Filopodia: the fingers that do the walking. Sci STKE. 2007;2007(400):re5.
16. Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, McCormick F, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84(5):723-34.
17. Miki H, Sasaki T, Takai Y, Takenawa T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature. 1998;391(6662):93-6.
18. Ma L, Rohatgi R, Kirschner MW. The Arp2/3 complex mediates actin polymerization induced by the small GTP-binding protein Cdc42. Proc Natl Acad Sci U S A. 1998;95(26):15362-7.
19. Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78(4):635-44.
20. Frugtniet B, Jiang WG, Martin TA. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer (Dove Med Press). 2015;7:99-109.
21. Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97(2):221-31.
22. Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A. Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol. 2001;11(21):1645-55.
23. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1(5):253-9.
24. Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17(3):139-52.
25. European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182-236.
26. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25-34.
27. Uka K, Aikata H, Takaki S, Shirakawa H, Jeong SC, Yamashina K, et al. Clinical features and prognosis of patients with extrahepatic metastases from hepatocellular carcinoma. World J Gastroenterol. 2007;13(3):414-20.
28. D'Avola D, Granito A, Torre-Alaez M, Piscaglia F. The importance of liver functional reserve in the non-surgical treatment of hepatocellular carcinoma. J Hepatol. 2022;76(5):1185-98.
29. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629-35.
30. Maldonado MDM, Medina JI, Velazquez L, Dharmawardhane S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front Cell Dev Biol. 2020;8:201.
31. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247-69.
32. Zhang X, Ren L, Wu J, Feng R, Chen Y, Li R, et al. ARHGEF37 overexpression promotes extravasation and metastasis of hepatocellular carcinoma via directly activating Cdc42. Journal of experimental & clinical cancer research : CR. 2022;41(1):230.
33. Murphy DA, Courtneidge SA. The 'ins' and 'outs' of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12(7):413-26.
34. Zeng Y, Guo Z, Hu Z, Liu M, Chen Y, Chen S, et al. FGD1 exhibits oncogenic properties in hepatocellular carcinoma through regulating cell morphology, autophagy and mitochondrial function. Biomed Pharmacother. 2020;125:110029.
35. Zhang J, Yang C, Gong L, Zhu S, Tian J, Zhang F, et al. RICH2, a potential tumor suppressor in hepatocellular carcinoma. Front Biosci (Landmark Ed). 2019;24(8):1363-76.
36. Tonucci FM, Almada E, Borini-Etichetti C, Pariani A, Hidalgo F, Rico MJ, et al. Identification of a CIP4 PKA phosphorylation site involved in the regulation of cancer cell invasiveness and metastasis. Cancer Lett. 2019;461:65-77.
37. Maldonado MDM, Dharmawardhane S. Targeting Rac and Cdc42 GTPases in Cancer. Cancer Res. 2018;78(12):3101-11.
38. Pelish HE, Peterson JR, Salvarezza SB, Rodriguez-Boulan E, Chen JL, Stamnes M, et al. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat Chem Biol. 2006;2(1):39-46.
39. Friesland A, Zhao Y, Chen YH, Wang L, Zhou H, Lu Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(4):1261-6.
40. Hong L, Kenney SR, Phillips GK, Simpson D, Schroeder CE, Noth J, et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J Biol Chem. 2013;288(12):8531-43.
41. Zins K, Gunawardhana S, Lucas T, Abraham D, Aharinejad S. Targeting Cdc42 with the small molecule drug AZA197 suppresses primary colon cancer growth and prolongs survival in a preclinical mouse xenograft model by downregulation of PAK1 activity. J Transl Med. 2013;11:295.
42. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33(31):4021-35.
43. Hussain NK, Jenna S, Glogauer M, Quinn CC, Wasiak S, Guipponi M, et al. Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat Cell Biol. 2001;3(10):927-32.
44. Aguilar BJ, Zhao Y, Zhou H, Huo S, Chen YH, Lu Q. Inhibition of Cdc42-intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol Ther. 2019;20(6):740-9.