Abstract
One of the mechanisms used by epigenetic therapy is the elevation of host cell-derived double stranded RNA (dsRNA) baseline levels through overexpression of genomic repetitive elements especially Alu retroelements. The dsRNAs trigger immunogenic responses since immune system cannot distinguish between endogenous and exogenous dsRNAs derived from viral infections; hence called “Viral mimicry response”. These dsRNAs are recognized by pattern recognition receptors (PRRs) such as MDA-5 which further induce inflammatory responses through interferon secretion. However, the response is limited through the function of some editing enzymes such as ADAR1 which destabilizes the formation of dsRNAs and renders the therapy less efficient through attenuating interferon secretion by immune cells. Since, some cancer cells can survive even after ADAR1 inhibition, it is speculated that there might be other mechanism which contribute to dsRNA destabilization. Since dsRNA formation derived from retroelement transcripts mimics viral infections, we tried to review the mechanistic approaches applied during host-pathogen interaction to highlight a possible candidate which might be cogitable for further investigations in epigenetic therapy. dsRNAs produced by RNA viruses are sensed by PRRs and activate nuclear factor erythroid 2 p45-related factor 2 (NRF2) which further downregulates STING protein and attenuates IFN release. RNA viruses such as SARS-CoV-2 have the potential to impair NRF2 signaling and eliminate its inhibitory effect from STING, leading to excessive release of IFNs and destroy pulmonary cells through cytokine release storm (CRS). Here, we briefly explain that NRF2, in a very downstream side of anti-viral response, might be a potential candidate target in combination with epigenetic therapy to circumvent the limitations in cancer epigenetic therapy.
Keywords
Epigenetic therapy, Viral mimicry, Interferon, Immunotherapy
Cancer and Immune Response
It is well established that cytotoxic CD8+ T cells play integral roles in eliminating malignant transformed cells. Immunotherapies based on T cells have been gathered increasing attention in cancer treatment. A series of conditions is required for immune response to tumor cells. First, tumor cells secrete tumor-associated antigens into the tumor microenvironment, which are then captured by antigen-presenting cell (APC) [1]. Antigen-loaded APCs process and present antigens through major histocompatibility (MHC) complexes to the cell surface and then transfer them to lymphoid organs. There, the selected peptide-MHC complexes are recognized by naïve T cells through T cell receptor (TCR). This induces the priming and activation of effector T cells. Passing through the circulating system, T cells recognize cancer cells by virtue of matching antigens in cancer cells recognized by TCR and peptide-MHC complexes. This recognition leads to cancer cell death by direct and indirect immune attack. A new round of anti-tumor immune response would be triggered through the release of additional antigens from dead tumor cells [2,3].
Role of Epigenetics in Cancer Therapy
One of the major mechanisms in cancer development and progression, is alteration in epigenome profile and aberrant DNA methylation [4-6]. Epigenetic modulation is a heritable DNA modification that changes the appearance of the chromatin window while maintains the DNA sequence intact, leading to gene expression regulation. Eukaryotic genes switch between "on" and "off" state by a variety of mechanisms [7,8]: chromatin remodeling; histone variant exchange; and the role of non-coding RNAs. Nucleosome distribution can be modified throughout the genome by moving the sites of packaged DNA [9,10]. The key regulator of chromatin structure is DNA methylation in CpG sites as well as post-translational modification of histones, including acetylation, methylation, and generalization. While, chromatin structure in the open state allows access of transcriptional activators, the closed state is associated with transcription [11].
Dysregulated patterns of DNA methylation are often associated to frequent mutations in genes that modulate DNA methylation such as DNMT3a and TET2 in human cancers, which leads to impaired gene expression. For example, local hypermethylation of tumor suppressor gene promoters suppresses their expression, which is directly linked with tumorigenesis [12].
Modification of various positions of histones attracted great attention for transcriptional regulation of genes in tumor cells. For example, silencing of repetitive DNA and transposons is highly related to H4K2me3. Loss of H4K20me3 is an important characterization of cancer. DNA methylation together with post-translational modification determine the transcriptional status which leads to tumor progression. In addition, epigenetics also affects anti-tumor immune responses, such as inducing neoantigen production, disrupting antigen presentation mechanisms, promoting inflammatory factor secretion and inducing immunosuppressive effects, thereby exacerbating tumor development [13].
DNA methyltransferase inhibitors (DNMTi) such as cytidine analogues 5-azacytidine (Azacytidine or AZA) and 5-aza-2´-deoxycytidine (Decitabine or DAC) have been synthesized in 1964 as first epigenetic drugs [14]. During DNA replication and transcription, DAC intercalates into DNA and AZA incorporates into both DNA and RNA. It was demonstrated that prolonged treatment with low-doses of AZA can eventually lead to the treatment of myeloplastic syndrome (MDS) [15,16].
Epigenetic Therapy and Immune Response
Immunotherapy meets epigenetic therapy through "viral mimicry" mechanism. This is an immune response induced by elevated levels of endogeneous nucleic acids often primed by overexpression of cellular retrotransposons such as long interspersed nuclear element (LINEs), Alu elements belonging to short interspersed nuclear element (SINEs) and long terminal repeat (LTR)/ endogenous retrovirus (ERVs) which form double stranded RNA (dsRNA) [17]. The ERVs represent a large fraction of repetitive elements in the human genome that are silenced by DNA methylation. Treatment with DNMTis allows cancer cells to enter a "viral mimicry" state in which they behave like virus-infected cells, leading to the activation of the interferon pathway [18].
Recent studies have reported increased levels of cytosolic double stranded RNA (dsRNA), higher than tolerable thresholds, as a cancer therapeutic strategy to promote antiviral responses [19]. For example, in ovarian cancer cell lines, DNMTis trigger the transcription of dsRNA by suppressing silent expression of hypermethylated ERVs. Upregulation of dsRNA activates cytoplasmic dsRNA sensors and downstream signaling pathways which induce interferon (INF)-β signaling [20]. The production of type I and type III IFNs induced by the viral mimicry pathway would increase antigen presentation and processing of cancer cells in the tumor microenvironment. The same finding is observed in colon cancer cells treated with 5-AZA-cdR.
Epigenetic Therapy and ADAR1 Dependency
Most prevalent RNA modification in mammalian cells is Adenosine to inosine (A-to-I) editing catalyzed by adenosine deaminase acting on the double-stranded RNA (ADAR) protein family. This kind of editing is currently known to be involved in the regulation of the immune system, protein recoding, microRNA biogenesis, RNA splicing, and heterochromatin formation. ADAR1 mediated editing occurs within dsRNAs, especially inverted Alu repeats, and is associated to variety of diseases including cancer, neurodegeneration, and metabolic disorders [21].
Epigenetic therapies such as AZA and DAC can perform their function through activating retroelements within human genomes. Studies have been demonstrated that the major source of epidrug-induced immunogenicity are inverted Alu repeats, mostly located downstream of ‘orphan’ CpG islands [22]. The transcript of retroelements can form dsRNA that activates PRRs such as (Melanoma differentiation-associated proteins (MDA-5) and RNA- dependent protein kinase (PKR) [18,20,23-26]. It was shown that upon epigenetic therapy, stimulated RNA sensors induce MAVs aggregation on the mitochondrial surface to activate TBK1 kinase which then phosphorylatesIRF-3/7 transcription factor, phosphorylated IRF3/7 dimerize and then translocate to nucleus to activate transcription o IFN genes [18,20]. This viral mimicry mechanism induces innate and adaptive immune responses leading to cancer cell suppression [27,28]. However, the clinical efficacy of cancer therapies against epigenetic repressors has been attenuated through ADAR1-based A-to-I editing system, destabilizing inverted repeat Alu dsRNAs [29].
Indeed, cancer cells hijack ADAR1-mediated dsRNA editing to circumvent innate immune responses such as interferon I and III secretion triggered by endogenous dsRNAs. ADAR1 disrupts the RNA duplex formation through dsRNA modification and diminishes sensing of dsRNA by PRRs [29]. Immunostimulatory dsRNA editing through ADAR1 reduces the double stranded to single stranded RNA ratio. This lower ratio of immunostimulatory dsRNAs sensed by PRRs leads to chronic versus acute IFN production. Cancer cells can escape chronic IFN response triggered by dsRNA. While chronic IFN production augments cancer aggressiveness, excessive or acute release of IFNs halts tumor progression [30]. Cancer cells can escape chronic IFN responses triggered by decreased ratio of dsRNAs. For example, global DNA hypomethylation of LINEs, SINEs, and LTR/ERVs in a subset of pancreatic ductal adenocarcinoma (PDAC) are associated with upregulated dsRNA expression, chronic IFN production accompanied by tumor metastasis and invasion [31].
However, survival of ADAR1-deficient cancer cells raises the question whether ADAR1 is the sole regulator of immune response after epigenetic therapy. Thus, if dsRNA formation cannot be prevented in the upstream, it is reasonable to ponder about manipulation of downstream responses after dsRNA formation and trigger, which leads to cancer invasiveness.
Double-edged Role of NRF2 in Cancer
NRF2 is a transcription factor which binds to various molecules such as small musculoaponeurotic fibrosarcoma (SMAF) proteins [32] or transcription factors c-JUN and JUND [33], to target antioxidant response elements (AREs), such as genes involved in cellular redox homeostasis, detoxification, metabolic balance, and macromolecular damage repair [34].
NRF2 has multifaceted roles in cancer cells. Although degradation process is the main process regulating NRF2 protein level, the control of NRF2 gene transcription is through its transcriptional start sites containing Myc and Jun binding sites [35]. Thus, the expression of NRF2 and its downstream genes can be augmented significantly by activating the oncogenic alleles of BRAF, C-MYC, and KRAS, reducing intracellular redox environment [36]. The oncogenic KRAS can activate MAPK-mediated NRF2 antioxidant function in pancreatic cancer [37]. Synthetic and extracted NRF2 activators such as curcumin, resveratrol, lycopene, and epigallocatechin-3-gallatecan promote antioxidant function of NRF2 and protect the cells from carcinogenic exposure [38-40].
On the other hand, NRF2 inhibitors such as brusatol, retinoic acid, and trigonelline can also be applied as anticancer agents. NRF2 inhibitors diminish drug detoxifying and cleaning enzymes, leading to cancer cell sensitization to chemotherapeutics [40-42].
NRF2 Anti-inflammatory Role
The activation of NRF2/ ARE signaling pathway plays a vital role in the escalation of chronic inflammation which contribute to cancer progression. ROS removal is thought to be the main molecular mechanism behind NRF2 mediated anti-inflammation as a result of upregulation of a large number of cytoprotective enzymes. NRF2 suppresses the activation of the pro-inflammatory genes and promotes the anti-inflammatory signaling [43].
Studies of putative viral cycle of RNA viruses [44] such as SARS-Cov-2 [45], highlights potential crosstalks with NRF2 activation. Upon viral infection, viral dsRNAs can inactivate eukaryotic initiation factor 2 (eIF2) through PKR and PKR-like endoplasmic reticulum kinase (PERK) which can be activated in response to viral infection [46]. PKR can activate autophagy cargo protein p62, which competes with NRF2 for binding KEAP1 [47] and induce autophagic degradation of KEAP1, an E3 ligase substrate adapter protein [48]. This leads to the activation of NRF2 transcriptional activity [45].
Under basal conditions, NRF2 interacts with KEAP1. This leads to the ubiquitination and further proteasomal degradation of NRF2, keeping the NRF2 level in a steady-state condition [34,49,50]. NRF2 is expressed at a steady-state level in the cell despite its high rate of turnover due to a time-elapsed interval (~15 min) between its biosynthesis and degradation. During this interval, NRF2 might transactivate its genes and accomplishes its role in driving their constitutive expression [51]. Viral nucleic acids inactivate KEAP1 through PKR [48]. Indeed, infected cells with viruses recognize viral nucleic acids by cytoplasmic and endosomal receptors, such as RIG-I [52], MDA5, PKR, and cGAS which signals through STING to induce an appropriate immune response. The activation of STING leads to the secretion of IFN I and III through interferon regulatory factor 3 (IRF3) [53]. NRF2 plays its immunoregulatory role by downregulation of STING, alleviating the excessive release of IFNs through STING activity leading to an appropriate immune response. RNA such as SARS-CoV2 (Figure 1) [54,55]. It was reported that lethal viruses have the ability to aberrate NRF2 axis and inhibits its function, leading to release of excessive IFNs, further CRS and irreversible damage to the host tissues [56].
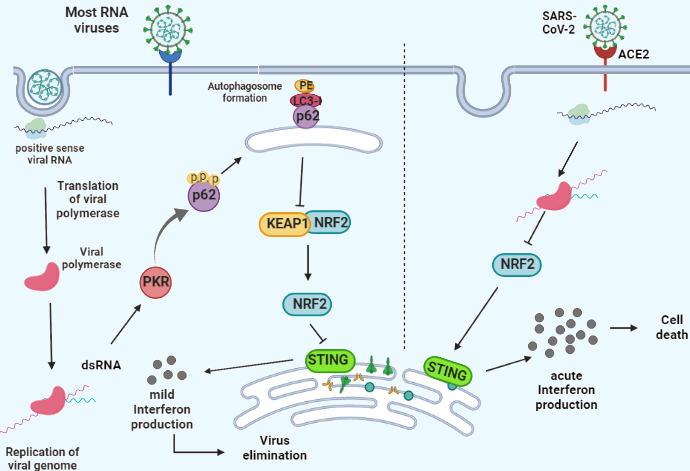
Figure 1. A scheme of RNA infection in host cell (Left). Viral RNA is sensed by PKR. PKR phosphorylates and activates p62 which is the scaffold for autophagosome formation, leading to autophagic degradation of KEAP. KEAP1 is NRF-2 inhibitor. The activation of NRF-2 downregulates the expression levels of STING, activation of which produces IFNs and immunogenic response. SARS-CoV-2 infection (Right) impairs NRF-2 signaling pathway. This results in the upregulation of STING (Left) and acute release of IFNs which leads to host cell death.
As mentioned earlier, chronic and alleviated release of IFNs can lead to tumor invasion while culling the cancer cell progression requires acute IFN production. The antitumorigenic as well as pro-tumorigenic role of STING has been reported previously in literature [57-60]. Furthermore, the key role of NRF2 in regulating STING has been established. Inhibiting NRF2 can eliminate its controlling effect from STING, allowing acute secretion of IFNs and further tumor rejection.
dsRNA, ADAR1 and NRF2 Connection
The mechanism by which elevated dsRNAs prime sublethal versus lethal IFN response requires further investigation. Gannon et al. demonstrated that an increased gene expression signature of interferons is predictive of ADAR1-dependency. They implied that despite previous studies, they did not observe any essential correlation between ADAR1 dependency and MDA5/MAVs signaling in lung cancer cell lines [61]. They reported that their data are distinct from previously published studies which demonstrated a critical role for MDA5/MAVs pathway in embryonic lethality phenotype found in Adar1-/- [62-64].
The question raised is whether the chronic exposure to exogenous viral infection leads to an immune exhaustion and reduces immune response [65]. Another explanation by Gannon et al., was that the downstream pathways that mediate cellular lethality after ADAR deletion may vary depending in the specific cell type, developmental stage and/or malignant nature of the cells under investigation. Additionally, there are several approaches which possibly can disrupt ADAR1 function in cancer cells such as direct enzymatic inhibition of its adenosine deaminase activity. Gannon et al. also reported that p150 isoform of ADAR1 prevents lethality in cancer cell lines at least partly through inhibition of the cytoplasmic RNA sensor PKR [61]. Although studies have shown ADAR1 deficiency decreases RNA editing and augments dsRNA stabilization, high levels of dsRNAs can activate PKR which in turn induce NRF2 activation. The inhibitory effect of NRF2 on STING may attenuate acute release of IFN I and III, which in turn prevent cancer cell death. This might be one reason why some cancer cells survive during epigenetic therapy even with ADAR1 loss of function. Since some studies have not found changes in MDA5/MAVs signaling pathway in cancer cells with ADAR deficiency, the PKR/NRF2/STING axis might be a landscape of more investigation during epigenetic therapy. Furthermore, the constitutive activation of NRF2 signaling pathway in a variety of cancers signifies the importance of this pathway during cancer epigenetic therapy [43].
For a therapy to be effective in cancer treatment it is critical to promote lethal acute immunogenicity rather than chronic immune response which results in cancer aggressiveness. Taking a deep look into mechanisms involved in RNA virus infections which are mostly fatal, can give us some hints to better understand the molecules involved in the crosstalk between epigenetic therapy and immune system and provide us with much comprehensive insight about limitation and the potential therapeutic strategies to overcome epigenetic therapy barricade. Some RNA viruses such as SARS-CoV-2 increase morbidity in patients through activation of CRS, leading to alveolar tissue damage through necrosis and apoptosis. They produce double stranded RNAs that are recognized by PRRs and activate the downstream NRF2 which downregulates STING and alleviates IFN release, leading to an appropriate inflammatory response against virus. However, RNA viruses apply their strategies to hijack STING signaling through suppression of NRF2 [66,67]. Removing NRF2 regulatory effect from STING can increase the magnitude of IFN release. The magnitude of IFN release is the challenging point in the cancer progression and therapy. While an acute IFN secretion leads to cancer cell death, the chronic and moderate IFN release results in cancer progression. In epigenetic therapy, when ADAR1 destabilizes dsRNA formation through its editing system, it can understimulate the PRRs sensing dsRNAs and keep NRF2 at cellular basal levels [51,63,68] and further attenuating pro-inflammatory responses through STING. However, ADAR1 knockdown, increase dsRNA formation which in turn activates NRF2 through PKR function, leading to STING downregulation and the following IFN I and III attenuation (Figure 2). Applying the strategy (ie. NRF2 impairment) that SARS-CoV-2 uses for inflammatory evasion and call/tissue damage through acute release of IFNs can be practical for tumor cell damage and loss of cancer cell fitness.
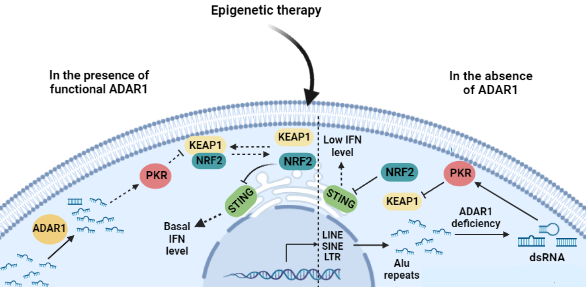
Figure 2. After epigenetic therapy, cancer cells use ADAR1 to destabilize dsRNA formation which understimulates PKR (Left). Since NRF2 has constitutive expression and there is a time interval between its biosynthesis and degradation, its steady-state levels can downregulate STING and IFNs are maintained at basal levels. ADAR1 knockout leads to the elevation in the dsRNA formation. The dsRNAs can fully-activate NRF2 through PKR activation, which in turn downregulates STING and lowers the release of IFNs (Right).
Conflicts of Interest
The author has no conflict of interest to disclose.
References
2. Fu C, Jiang A. Dendritic cells and CD8 T cell immunity in tumor microenvironment. Frontiers in Immunology. 2018 Dec 20;9:3059.
3. Zhang Y, Du X, Liu M, Tang F, Zhang P, Ai C, et al. Hijacking antibody-induced CTLA-4 lysosomal degradation for safer and more effective cancer immunotherapy. Cell Research. 2019 Aug;29(8):609-27.
4. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature Reviews Genetics. 2002 Jun 1;3(6):415-28.
5. Esteller M. Epigenetics in cancer. New England Journal of Medicine. 2008 Mar 13;358(11):1148-59.
6. Fardi M, Solali S, Hagh MF. Epigenetic mechanisms as a new approach in cancer treatment: An updated review. Genes & Diseases. 2018 Dec 1;5(4):304-11.
7. Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007 Feb 23;128(4):683-92.
8. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013 Mar 28;153(1):38-55.
9. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001 Aug 10;293(5532):1068-70.
10. Kouzarides T. Chromatin modifications and their function. Cell. 2007 Feb 23;128(4):693-705.
11. Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007 Feb 23;128(4):707-19.
12. Schübeler D. Function and information content of DNA methylation. Nature. 2015 Jan 15;517(7534):321-6.
13. Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 2005 Apr;37(4):391-400.
14. Šorm F, Piskala A, Čihák A, Veselý J. 5-Azacytidine, a new, highly effective cancerostatic. Experientia. 1964 Apr;20:202-3.
15. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980 May 1;20(1):85-93.
16. Jones PA, Taylor SM. Hemimethylated duplex DNAs prepared from 5-azacytidine-treated cells. Nucleic Acids Research. 1981 Jun 25;9(12):2933-47.
17. Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Molecular Pharmacology. 2004 Jan 1;65(1):18-27.
18. Roulois D, Yau HL, Singhania R, Wang Y, Danesh A, Shen SY, et al, Pugh TJ, O’Brien C. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015 Aug 27;162(5):961-73.
19. Ishak CA, Classon M, De Carvalho DD. Deregulation of retroelements as an emerging therapeutic opportunity in cancer. Trends in Cancer. 2018 Aug 1;4(8):583-97.
20. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015 Aug 27;162(5):974-86.
21. Shevchenko G, Morris KV. All I's on the RADAR: role of ADAR in gene regulation. FEBS Letters. 2018 Sep;592(17):2860-73.
22. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes & Development. 2011 May 15;25(10):1010-22.
23. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. 2017 Aug 24;548(7668):471-5.
24. Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell. 2018 Jul 26;174(3):549-63.
25. Cuellar TL, Herzner AM, Zhang X, Goyal Y, Watanabe C, Friedman BA, et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. Journal of Cell Biology. 2017 Nov 6;216(11):3535-49.
26. Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, Cheung TK, et al. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell. 2017 Aug 14;32(2):221-37.
27. Yau HL, Ettayebi I, De Carvalho DD. The cancer epigenome: exploiting its vulnerabilities for immunotherapy. Trends in Cell Biology. 2019 Jan 1;29(1):31-43.
28. Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nature Reviews Cancer. 2019 Mar;19(3):151-61.
29. Levanon EY, Eisenberg E, Yelin R, Nemzer S, Hallegger M, Shemesh R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nature Biotechnology. 2004 Aug 1;22(8):1001-5.
30. Boukhaled GM, Harding S, Brooks DG. Opposing roles of type I interferons in cancer immunity. Annual Review of Pathology: Mechanisms of Disease. 2021 Jan 24;16:167-98.
31. Espinet E, Gu Z, Imbusch CD, Giese NA, Büscher M, Safavi M, et al. Aggressive PDACs Show Hypomethylation of Repetitive Elements and the Execution of an Intrinsic IFN Program Linked to a Ductal Cell of OriginAutonomous IFN Program in Aggressive Ductal-Derived PDAC. Cancer Discovery. 2021 Mar 1;11(3):638-59.
32. Otsuki A, Yamamoto M. Cis-element architecture of Nrf2–sMaf heterodimer binding sites and its relation to diseases. Archives of Pharmacal Research. 2020 Mar;43:275-85.
33. Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene. 1998 Dec;17(24):3145-56.
34. Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nature Reviews Drug Discovery. 2019 Apr;18(4):295-317.
35. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011 Jul 7;475(7354):106-9.
36. Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, Jeddi F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Communication and Signaling. 2022 Jun 30;20(1):100.
37. Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Frontiers in Physiology. 2013 Sep 12;4:246.
38. Milkovic L, Zarkovic N, Saso L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biology. 2017 Aug 1;12:727-32.
39. Robledinos-Antón N, Fernández-Ginés R, Manda G, Cuadrado A. Activators and inhibitors of NRF2: a review of their potential for clinical development. Oxidative Medicine and Cellular Longevity. 2019 Jul 14;2019.
40. Sova M, Saso L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug design, Development and Therapy. 2018 Sep 25:3181-97.
41. Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1–Nrf2 system in cancers: stress response and anabolic metabolism. Frontiers in Oncology. 2012 Dec 26;2:200.
42. Telkoparan-Akillilar P, Panieri E, Cevik D, Suzen S, Saso L. Therapeutic targeting of the NRF2 signaling pathway in cancer. Molecules. 2021 Mar 5;26(5):1417.
43. Wu S, Lu H, Bai Y. Nrf2 in cancers: A double-edged sword. Cancer Medicine. 2019 May;8(5):2252-67.
44. Chen YG, Hur S. Cellular origins of dsRNA, their recognition and consequences. Nature Reviews Molecular Cell Biology. 2022 Apr;23(4):286-301.
45. Cuadrado A, Pajares M, Benito C, Jiménez-Villegas J, Escoll M, Fernández-Ginés R, et al. Can activation of NRF2 be a strategy against COVID-19?. Trends in Pharmacological Sciences. 2020 Sep 1;41(9):598-610.
46. Krähling V V, Stein DA, Spiegel M, Weber F, Mühlberger E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. Journal of Virology. 2009 Mar 1;83(5):2298-309.
47. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology. 2010 Mar;12(3):213-23.
48. Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proceedings of the National Academy of Sciences. 2012 Aug 21;109(34):13561-6.
49. Cuadrado A, Manda G, Hassan A, Alcaraz MJ, Barbas C, Daiber A, et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: a systems medicine approach. Pharmacological Reviews. 2018 Apr 1;70(2):348-83.
50. Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends in Biochemical Sciences. 2014 Apr 1;39(4):199-218.
51. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. Journal of Biological Chemistry. 2009 May 15;284(20):13291-5.
52. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. Journal of Virology. 2008 Jan 1;82(1):335-45.
53. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011 Oct 27;478(7370):515-8.
54. Olagnier D, Lababidi RR, Hadj SB, Sze A, Liu Y, Naidu SD, et al. Activation of Nrf2 signaling augments vesicular stomatitis virus oncolysis via autophagy-driven suppression of antiviral immunity. Molecular Therapy. 2017 Aug 2;25(8):1900-16.
55. Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nature Communications. 2018 Aug 29;9(1):3506.
56. Deramaudt TB, Dill C, Bonay M. Regulation of oxidative stress by Nrf2 in the pathophysiology of infectious diseases. Médecine Et Maladies Infectieuses. 2013 Mar 1;43(3):100-7.
57. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung CancerSilencing of STING in KRAS-LKB1–Mutant Lung Cancer. Cancer Discovery. 2019 Jan 1;9(1):34-45.
58. Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metabolism. 2015 Jan 6;21(1):81-94.
59. Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proceedings of the National Academy of Sciences. 2017 Oct 24;114(43):E9066-75.
60. Kwon J, Bakhoum SF. The cytosolic DNA-sensing cGAS–STING pathway in cancer. Cancer Discovery. 2020 Jan;10(1):26-39.
61. Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nature Communications. 2018 Dec 21;9(1):5450.
62. Heraud-Farlow JE, Walkley CR. The role of RNA editing by ADAR1 in prevention of innate immune sensing of self-RNA. Journal of Molecular Medicine. 2016 Oct;94:1095-102.
63. Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Reports. 2014 Nov 20;9(4):1482-94.
64. Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity. 2015 Nov 17;43(5):933-44.
65. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. 2013 Apr 12;340(6129):202-7.
66. Sun L, Xing Y, Chen X, Zheng Y, Yang Y, Nichols DB, et al. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PloS One. 2012 Feb 1;7(2):e30802.
67. Chen X, Yang X, Zheng Y, Yang Y, Xing Y, Chen Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein & Cell. 2014 May;5(5):369-81.
68. Nallagatla SR, Toroney R, Bevilacqua PC. A brilliant disguise for self RNA: 5'-end and internal modifications of primary transcripts suppress elements of innate immunity. RNA Biology. 2008 Jul 1;5(3):140-4.