Abstract
Alzheimer disease (AD) is recognized by a gradual loss of memory which never returns to normal, called dementia caused due to the death of functional neural cells. Only 10-15% cases are found genetically involved. However, in all the cases of AD, beta amyloid (Aβ) protein forms plaques, and the tau protein forms tangles which disrupts the normal neuronal functions for transporting neurotransmitters, nutrient, and also communication with other neural cells; ultimately develops the disease. We have reviewed here the presently available all the treatment options, and discussed, not only why but how, a better option can be developed for AD treatment.
Keywords
Alzheimer’s Disease, Amyloid β-protein (Aβ), Tau protein, Neuron; Cell therapy, Gene therapy
Introduction
Alzheimer’s disease (AD)
It is one of the most prevalent neurodegenerative disorder in the world, first described by a German Scientist, Alois Alzheimer, in 1906. The main symptom of the disease is dementia, a steady loss of memory; and as the disease progress ability to speak, to think and to do daily activities decline [1].
The early-onset of AD generally starts before 55 years of age, and late-onset of AD, generally starts after 60 years of age [2].
Prevalence
Approximately 5.5 million people in the United States and 47 million people worldwide are currently affected by AD [2,3]. It is expected that by 2050, nearly a million new cases per year may develop [4].
Symptoms: What are the symptoms of AD
Memory loss is usually the first sign of AD which is different than normal memory problems. Unlike in other cases where one can forget something for the time being but can recollect later, AD person’s loss of memory is irreversible.
Followings are the symptoms associated with mild, moderate, and severe AD. Symptoms vary as the disease progresses (Figure 1).
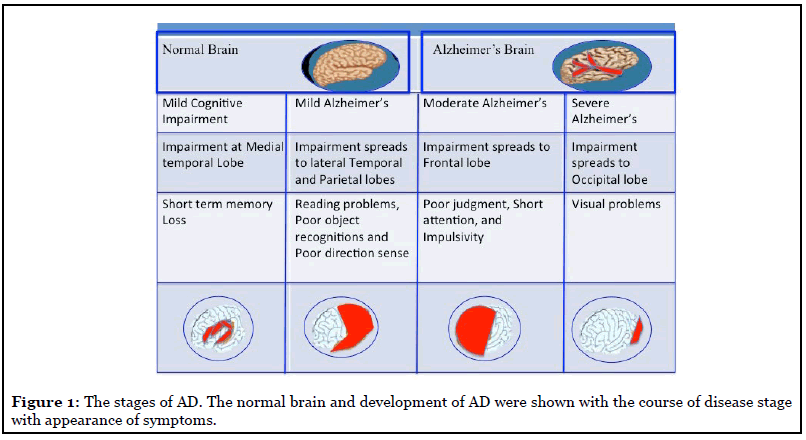
Mild AD: Usually, a person with mild AD avoids new and unfamiliar situations; has trouble learning and remembering new information, speaks more slowly than in the past; makes wrong decisions; may have mood swings and become depressed, grouchy, or restlessness [5].
Moderate AD: A person in this case typically has problems recognizing close friends and family members; becomes more restless, especially in late afternoon and at night; has problems in reading, writing, and dealing with numbers; has trouble in taking decisions, communication; and has problem to find the known places [5].
Severe AD: With severe AD, a person usually cannot do any personal daily work without help. They have trouble with body balance, may fall often during walking. They lose bowel or bladder control [5].
Other conditions: Other conditions may include hyperthyroidism or hypothyroidism; depression; and may have kidney disease, liver disease; sometimes found association with HIV (human immunodeficiency virus) infections [6,7].
Causes of AD
Sporadic: AD pathology consists of amyloid-β (Aβ) deposition in the brain, the hyperphosphorylation of tau proteins, and neuroinflammation through glial activation [8]. Aβ peptides are often referred to by the length of their amino acid sequences and can be found in cerebral and peripheral tissues. Although there are many conformations of Aβ, which commonly consists of 36–43 amino acids, Aβ42 is known to aggregate the most readily and aid in the formation of neuritic plaques [9]. Studying the aggregated forms of hyperphosphorylated tau, also referred to as neurofibrillary tangles, can determine the extent of brain and nerve damage exhibited by the AD patients.
In healthy neurons, tau protein normally binds to and stabilizes microtubules which help nutrients and molecules to be transported from the cell body to the axon and dendrites [10,11], but in all the cases of AD, beta amyloid (Aβ) protein plaques and the tau protein tangles disrupts those normal neuronal functions and communication with the other neural cells; and ultimately develops the disease [2,12-14] (Figure 2). However, AD therapeutics based on the amyloid hypothesis have repeatedly failed in clinical trials [15,16].
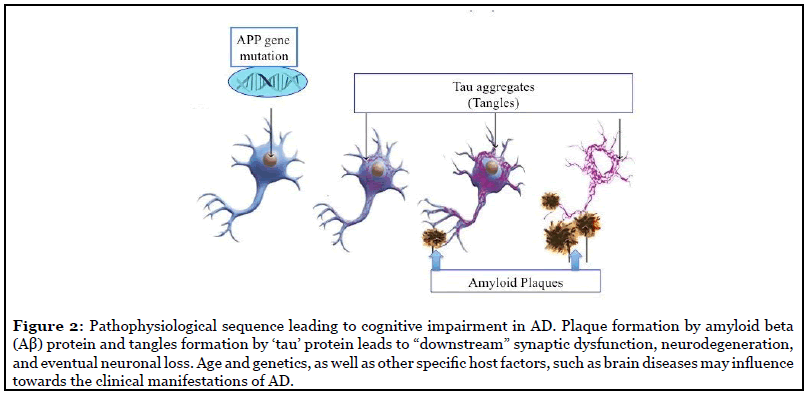
Involvement of Gene(s): Normally, a gene TREM2 is involved in microglia cells and astrocytes to clear betaamyloid protein plaques from the brain and helps to reduce the inflammation in the brain [17]. Inactivity of this gene/gene-product causes the plaque formation between neurons. Emerging evidence suggests that Alzheimer’srelated memory loss results from a complex interplay among abnormal accumulation of tau and β-amyloid proteins and several other factors [18].
Besides, identification of e4 allele of apolipoprotein E (ApoE) have been found in some AD families conferring the strongest genetic risk identified for AD development [3]. One in five people carries this allele, and carriers are three times more likely than non-carriers to develop AD in future [3]. ApoE plays a role in the processing and clearing the amyloid precursor protein (APP), and it is thought that carriers of the e4 allele are unable to effectively clear APP, leading to increased production and deposition of betaamyloid.
However, only 10-15% of cases are genetic, and the vast majority of cases are sporadic and attributed to many risk factors. During the normal aging process, production of plaques and tangles occurs. Once formed, plaques and tangles cause more inflammation, more formation of additional plaques and tangles ultimately leading to cognitive loss [3,12].
In some meta-analysis it has been found that approximately one-third of AD cases worldwide may be related to some risk factors, like diabetes mellitus, hypertension, obesity, physical inactivity, depression, smoking [19]. It may be possible to prevent or delay the onset of AD by controlling these risk factors.
Diagnosis of AD
AD is diagnosed generally after other conditions are ruled out [20].
Initial tests: Past health, physical exam to find out any physical problem, and mental health assessment are the routine initial checkup.
Lab tests: General lab tests for certain minerals or chemicals level in the blood; liver disease; abnormal thyroid levels; or nutritional problems, such as folic acid or vitamin B12 deficiencies.
Imaging and other tests:
• Brain imaging tests; an MRI (Magnetic Resonance Imaging) of the head; positron emission tomography (PET); and single photon emission tomography (SPECT); an electroencephalogram (EEG).
• A lumbar puncture to test for certain proteins in the spinal fluid.
Treatment options of AD
There is no cure yet for AD. More than 250 clinical trials have been conducted on a variety of treatments, including several that are capable of removing Aβ plaques from the brain. All have failed to reverse the progression of the disease [21].
Inhibition of enzymatic breakdown of acetylcholine: Acetylcholine, an important neurotransmitter is responsible for memory and cognitive function. Prevention of its enzymatic breakdown by acetylcholinesterase (AChE) inhibitors, such as donepezil, rivastigmine, and galantamine are approved to treat dementia in patients with AD [22]. However, these drugs may cause bradycardia and orthostatic hypotension, and also diarrhea, vomiting, nausea, fatigue, insomnia, loss of appetite, and weight loss [23].
All the chemo inhibitors of different enzymes related to AD development may slow down the disease progression but they do not significantly improve cognitive function or cure the disease, and therefore the clinical significance of their effectiveness is questionable [24].
Current research and possible new agents. In 2018, the AD drug-development pipeline includes 112 agents in phase I, II, or III trials; 63% of them are diseasemodifying therapies aimed at changing the course of AD and improving outcomes rather than managing symptoms [25]. About one-quarter of the drugs in development are for testing their ability to enhance cognition; and 10% of the drugs are intended to decrease behavioral symptoms such as agitation, apathy, and sleep disturbances [25,26].
Secretase inhibitors: Inhibition of the secretase enzymes involved in generating beta-amyloid protein from APP, have demonstrated the ability to reduce the formation of beta-amyloid plaques, but they have not been shown to reverse existing plaques or improve cognition [27,28].
Inhibition of tau protein: Protein tau is associated with neurofibrillary tangle formation [23]. Initial studies for reduction of tau aggregation, were not successful, however, new strategies are being tested on seven tau immunotherapies in phase I and II trials [25].
NMDA (N-methyl-D-aspartate) receptor antagonists: NMDA receptor allows glutamate to connect to a cell and allows calcium influx into the cell, for carrying the electrical or chemical signal. This is important for learning and memory. However, in AD, the nerve cells get too much glutamate, and that can damage to them. NMDA receptor antagonists make it harder for glutamate to “dock”, but they still let important signals to flow between the cells. NMDA receptor antagonist Memantine (Namenda XR), has been approved in the U.S. and Europe as a treatment for AD [29].
Inhibition of serotonin uptake: Given the failure to develop disease-modifying therapies for AD, strategies aiming at preventing or delaying the onset of the disease are being prioritized. Data from animal studies have shown a beneficial impact of selective serotonin reuptake inhibitors on pathophysiological biomarkers of AD including amyloid burden, tau deposits and neurogenesis. In humans, studies focusing on subjects with a prior history of depression also showed a delay in the onset of AD in those treated with most selective serotonin reuptake inhibitors. However, being noticed the methodological heterogeneity between studies, it is thus needed to conduct a large scale randomized controlled trials with long follow up periods to clarify the dose-effect relationship of specific serotonergic antidepressants on AD prevention [30].
Antioxidants: Free radicals tend to build up in nerve cells as we get older, and that oxidative stress can cause AD [31]. There are a lot of different antioxidants, like betacarotene, vitamins C and E, and resveratrol, which are used for Alzheimer’s treatment [32,33]. The antioxidant connection is a hot area in Alzheimer’s research, but everyone agrees that more still needs to be done.
Supplements: Some people have tried alternative remedies, including coenzyme Q10, coral calcium, huperzine A, and omega-3 fatty acids to prevent or treat AD. There’s not yet enough research to show if they do or do not work [34].
Further, a mechanism-based treatment for AD were also discussed elsewhere [35].
Possibilities of gene therapy: Compelling evidence from animal studies suggests that neurotrophic factors can potentially halt the progression of neuro-degeneration in diseases such as AD, resulting in improved motor and cognitive performance [36-38].
Delivery of neurotrophic factors to the CNS is a challenge since most proteins do not cross the blood–brain barrier, and systemic injection of certain growth factors results in strong peripheral side effects. Viral vector-mediated gene transfer allows for long-term and local expression of a foreign gene after a single injection [38-40].
NGF Gene Delivery: A first Phase I clinical trial with NGF gene therapy in AD has been completed in 2005. Fibroblasts of the patients were genetically modified to express NGF and were grafted into the nucleus [41-44]. PET studies suggest that the cholinergic projections to the cortex are re-activated by NGF [45].
Secondly, a decrease in disease progression was presented based on cognition assessment with the use of the Mini- Mental State Examination scores [45]. No negative effects of NGF administration (weight loss or pain) were noted in the AD patients, even up to 2 years post implantation. However, it was also reported earlier that NGF can induce β-amyloid precursor protein, and therefore may accelerate the pathology of AD instead [46].
Prevention of AD
Most of the AD (99% of all cases) really can be prevented by implementing specific lifestyle, changes and taking certain dietary supplements, which are most effective when combined together into a daily action plan [47].
Older adults who stay mentally active may be at lower risk for AD. Reading, playing cards and other games, working crossword puzzles, and even watching television or listening to the radio may help them to avoid symptoms of the disease [48].
Miscellaneous efforts to manage the AD
In addition to conventional treatment of AD, there are different types of therapies and counseling, that may help Alzheimer’s patient to help the management of symptoms in their daily life.
Occupational therapy: An occupational therapist can evaluate one’s strengths, weaknesses, and ability to understand where the AD patient needs help, and also a therapist can help the AD patient to improve some of his physical skills [49].
Physical therapy: Physical therapists can help AD patient to improve his balance and lower his chances of falling. The exercise can also be good for his mood [50].
Art and music therapies: Some science shows that these treatments, which stimulate the senses, can improve mood, behavior, and day-to-day function for people with Alzheimer’s. Art and music may help trigger memories and help people reconnect with the world around them. Painting, drawing, and other forms of art therapy can help people with Alzheimer’s disease express themselves. Expression through art can become especially important as a person’s ability to communicate through words deteriorates [51].
Massage: This hands-on therapy may help in two ways. It can ease agitation and may improve sleep [52].
Pet therapy: People who used to enjoy being with pets may find contact with them enriching or soothing [53].
Dealing with previous hobbies. Pursuing hobbies or interests that used to be familiar can help a person with Alzheimer’s feel more stable about their lives, like gardening, cooking, or any other activity that the person used to enjoy [54].
New possibilities with cell therapy
Regenerative cell therapy could be a promising approach for replacement of the loss of neurons and synapses in AD due to the accumulation of both amyloid plaques and neurofibrillary tau tangles (Figure 3) [55].
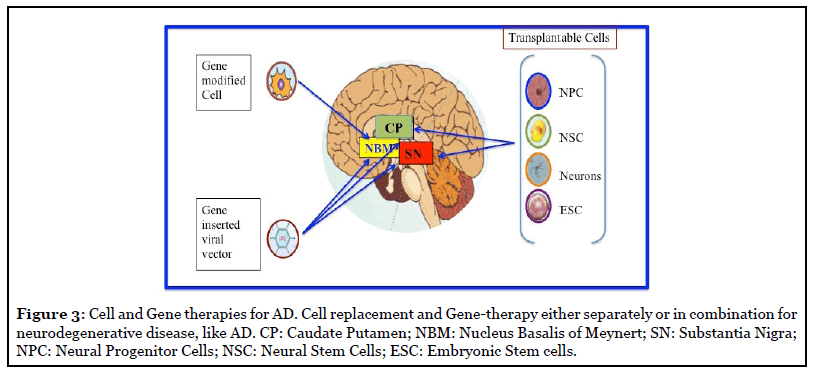
Stem cell Strategy: Stem cells have high migratory capacity after transplantation into the brain, and can be genetically modified in vitro. Therefore, they can be an efficient candidate to delivery neurotrophic factors or enhance gene expression to modify the course of the disease [56]. However, it has not yet been determined whether the curative effect depends directly on the promotion of neuronal regeneration by NSCs, or whether accompanying events, such as enhanced glial regeneration and other types of trophic support, are more important.
In the Aβ-induced AD model, the transplantation of ESCsderived NPCs into the injured hippocampus demonstrate the formation of synapses between host and grafted neural cells, and also improve the memory dysfunction of the AD models [57,58]. Neurons derived from human induced pluripotent stem cells (iPSCs) are endowed with a remarkable potential to establish orthotopic long-range projections in the adult mammalian brain [59]. Meanwhile, in another mouse AD model, the transplantation of NSCs was reported to improve cognition function mediated by the neurotrophic factor BDNF [60].
However, care should be taken with the characterization of these cells in regard to their multipotentiality and genetic stability with increasing passages in culture, as transformed cells may contribute to the formation of tumors [61], and also face some ethical issues [62].
Selection of right Cells, and its modifications: Human Neural stem cells (hNSCs) which are efficient in production and release of Dopamine, and neurofactors BDNF and GDNF, can be considered for PD cell therapy [63]. The most important physiological aspect of functional hNSCs is their capability to synthesize DA and also to catabolize any excess DA which is neurotoxic, and thereby maintain their physiologic label [64] However, hNSCs are slow growing cells and senesce after a few passages rendering a low level of supply for treatment. Attempts are taken in our lab to develop a natural cell modification method to increase the growth potential and survival length of hNSCs by a cell-cell interaction using melanocyte as a partner cell (personal communication).
Conclusions
It has been a century since the first description of AD was made, however, no proven effective treatment is available to date which can either delay the onset or slow the progression of AD. A combination of psychosocial, behavioral, and pharmacologic strategies aim to slow the process of AD and preserving quality of life as long as it is possible.
Although stem cell-based replacement strategies carried out in animal models have shown promising results, there are still many hurdles to overcome before these approaches can be translated into the AD patients. One major challenge is the development of a safe method to deliver stem cells to the injury region. In addition, the stage of differentiation of those cells needs careful consideration: fully differentiated cells are associated with a smaller efficiency due to poor viability, while undifferentiated cells present a higher risk of undirected differentiation and uncontrolled proliferation.
The development of novel cells, and gene-based therapies, or perhaps a combination of the two, could be beneficial in genuine repair and rescue of degenerating Dopaminergic (DAergic) and cholinergic neurons in the future (Figure 3).
Abbreviations
AD: Alzheimer’s Disease; SN: Substantia Nigra; APP: Amyloid Precursor Protein; Aβ: amyloid β-protein; iPSCs: induced Pluripotent Stem Cells; NGF : Nerve Growth Factor; Tau protein: τ proteins; PET: Positron Emission Tomography; SPECT: Single Photon Emission Tomography; ApoE4: e4 allele of Apolipoprotein E; NSC: Neural Stem Cells; NPC: Neural Progenitor Cells; ESC: Embryonic Stem Cells; EEG: Electroencephalogram; CT scan: Computed Tomography scan; MRI: Magnetic Resonance Imaging AChE: Acetylcholinesterase; DAergic: DOPAMINergic
Acknowledgment
We acknowledge all our staff members, scientists from AllExcel, Inc. for their support during the writing of this review by providing materials and editing.
Authors Contribution
Both the authors contributed equally.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
References
2. Wint D. Alzheimer’s disease. www.clevelandclinicmeded. com/medicalpubs/diseasemanagement/neurology/ alzheimers-disease.
3. dos Santos P, Leide C, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA,et al. Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Current Medicinal Chemistry. 2018 Aug 1;25(26):3141-59.
4. Kumar A, Singh A. A review on Alzheimer’s disease pathophysiology and its management: an update. Pharmacological Reports. 2015 Apr 1;67(2):195-203.
5. Förstl H, Kurz A. Clinical features of Alzheimer’s disease. European archives of Psychiatry and Clinical Neuroscience. 1999 Dec 1;249(6):288-90.
6. Basu G, Mohapatra A. Interactions between thyroid disorders and kidney disease. Indian Journal of Endocrinology and Metabolism. 2012 Mar;16(2):204.
7. Clifford DB, Ances BM. HIV-associated neurocognitive disorder. The Lancet infectious diseases. 2013 Nov 1;13(11):976-86.
8. Blennow K, Zetterberg H, Fagan AM. Fluid biomarkers in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2012 Sep 1;2(9):a006221.
9. Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harbor Perspectives in Medicine. 2012 Jun 1;2(6):a006262.
10. Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Current Alzheimer Research. 2010 Dec 1;7(8):656-64.
11. Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, et al. Role of Tau as a microtubule associated protein: structural and functional aspects. Frontiers in Aging Neuroscience. 2019;11:204.
12. Agamanolis DP. 33 Disorders of Amino Acid Metabolism and Canavan Disease. Developmental Neuropathology. 2018 Feb 26:403.
13. Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clinical interventions in aging. 2007 Sep;2(3):347.
14. Massoud F, Gauthier S. Update on the pharmacological treatment of Alzheimer’s disease. Current neuropharmacology. 2010 Mar 1;8(1):69-80.
15. Mullane K, Williams M. Alzheimer’s disease beyond amyloid: Can the repetitive failures of amyloid-targeted therapeutics inform future approaches to dementia drug discovery?. Biochemical pharmacology. 2020 Jul 1;177:113945.
16. Gold M. Phase II clinical trials of anti–amyloid β antibodies: When is enough, enough?. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2017 Sep 1;3(3):402-9.
17. Gratuze M, Leyns CE, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Molecular Neurodegeneration. 2018 Dec;13(1):1-6.
18. Rajmohan R, Reddy PH. Amyloid-beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. Journal of Alzheimer’s Disease. 2017 Jan 1;57(4):975-99.
19. Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. The Lancet Neurology. 2014 Aug 1;13(8):788-94.
20. Neugroschl J, Wang S. Alzheimer’s disease: diagnosis and treatment across the spectrum of disease severity. Mount Sinai Journal of Medicine: A Journal of Translational and Personalized Medicine. 2011 Jul;78(4):596-612.
21. Grill JD, Cummings JL. Current therapeutic targets for the treatment of Alzheimer’s disease. Expert review of Neurotherapeutics. 2010 May 1;10(5):711-28.
22. H Ferreira-Vieira T, M Guimaraes I, R Silva F, M Ribeiro F. Alzheimer’s disease: targeting the cholinergic system. Current Neuropharmacology. 2016 Jan 1;14(1):101-15.
23. Isik AT, Soysal P, Stubbs B, Solmi M, Basso C, Maggi S, et al. Cardiovascular outcomes of cholinesterase inhibitors in individuals with dementia: a meta-analysis and systematic review. Journal of the American Geriatrics Society. 2018 Sep;66(9):1805-11.
24. Winslow BT, Onysko M, Stob CM, Hazlewood KA. Treatment of Alzheimer disease. American Family Physician. 2011 Jun 15;83(12):1403-12.
25. Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s & Dementia: Translational Research & Clinical Interventions. 2018 Jan 1;4:195-214.
26. Frozza RL, Lourenco MV, De Felice FG. Challenges for Alzheimer’s disease therapy: insights from novel mechanisms beyond memory defects. Frontiers in Neuroscience. 2018 Feb 6;12:37.
27. Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, et. al. Alzheimer’s Disease. Lancet. 2016; 388: 505-517.
28. Moussa CE. Beta-secretase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opinion on Investigational Drugs. 2017 Oct 3;26(10):1131- 6.
29. Olivares D, K Deshpande V, Shi Y, K Lahiri D, H Greig N, T Rogers J, et al. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Current Alzheimer Research. 2012 Jul 1;9(6):746-58.
30. Mdawar B, Ghossoub E, Khoury R. Selective serotonin reuptake inhibitors and Alzheimer’s disease. Neural Regeneration Research. 2020 Jan;15(1):41.
31. Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Current Neuropharmacology. 2009 Mar 1;7(1):65- 74.
32. Aliev G, Obrenovich ME, Reddy VP, Shenk JC, Moreira PI, Nunomura A, et al. Antioxidant therapy in Alzheimer’s disease: theory and practice. Mini Reviews in Medicinal Chemistry. 2008 Nov;8(13):1395.
33. Sawda C, Moussa C, Turner RS. Resveratrol for Alzheimer’s disease. Annals of the New York Academy of Sciences. 2017 Sep;1403(1):142.
34. https://www.alz.org/alzheimers-dementia/treatments/ alternative-treatments
35. Davies P, Koppel J. Mechanism-based treatments for Alzheimer’s disease. Dialogues in Clinical Neuroscience. 2009 Jun;11(2):159
36. Kordower JH. In vivo gene delivery of glial cell line–derived neurotrophic factor for Parkinson’s disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2003;53(S3):S120-34.
37. Tuszynski MH, Alksne J, Bakay RA, Pay MM, Merrill D, Thal LJ. Growth factor gene therapy for Alzheimer disease. Neurosurgical Focus. 2002 Nov 1;13(5):1-5.
38. Dass B, Olanow CW, Kordower JH. Gene transfer of trophic factors and stem cell grafting as treatments for Parkinson’s disease. Neurology. 2006 May 23;66(10 suppl 4):S89-103.
39. Hendriks WT, Ruitenberg MJ, Blits B, Boer GJ, Verhaagen J. Viral vector-mediated gene transfer of neurotrophins to promote regeneration of the injured spinal cord. Progress in Brain Research. 2004 Jan 1;146:451-76.
40. Blesch A, Conner JM, Tuszynski MH. Modulation of neuronal survival and axonal growth in vivo by tetracyclineregulated neurotrophin expression. Gene Therapy. 2001 Jun;8(12):954-60.
41. Tuszynski MH, Smith DE, Roberts J, McKay H, Mufson E. Targeted intraparenchymal delivery of human NGF by gene transfer to the primate basal forebrain for 3 months does not accelerate β-amyloid plaque deposition. Experimental Neurology. 1998 Dec 1;154(2):573-82.
42. Tuszynski MH, Roberts JA, Senut MC, Gage FH. Gene therapy in the adult primate brain: intraparenchymal grafts of cells genetically modified to produce nerve growth factor prevent cholinergic neuronal degeneration. Gene Therapy. 1996 Apr;3(4):305-14.
43. Chen KS, Gage FH. Somatic gene transfer of NGF to the aged brain: behavioral and morphological amelioration. Journal of Neuroscience. 1995 Apr 1;15(4):2819-25.
44. Martinez-Serrano A, Fischer W, Björklund A. Reversal of age-dependent cognitive impairments and cholinergic neuron atrophy by NGF-secreting neural progenitors grafted to the basal forebrain. Neuron. 1995 Aug 1;15(2):473-84.
45. Tuszynski MH, Thal L, Pay M, Salmon DP, Bakay R, Patel P, et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nature Medicine. 2005 May;11(5):551-5.
46. Mobley WC, Neve RL, Prusiner SB, McKinley MP. Nerve growth factor increases mRNA levels for the prion protein and the beta-amyloid protein precursor in developing hamster brain. Proceedings of the National Academy of Sciences. 1988 Dec 1;85(24):9811-5.
47. Isik AT. Late onset Alzheimer’s disease in older people. Clinical Interventions in Aging. 2010;5:307.
48. Schultz SA, Larson J, Oh J, Koscik R, Dowling MN, Gallagher CL, et al. Participation in cognitively-stimulating activities is associated with brain structure and cognitive function in preclinical Alzheimer’s disease. Brain Imaging and Behavior. 2015 Dec 1;9(4):729-36.
49. Piersol CV, Jensen L, Lieberman D, Arbesman M. Occupational therapy interventions for people with Alzheimer’s disease. American Journal of Occupational Therapy. 2018 Jan 1;72(1):7201390010p1-6.
50. Teri L, Gibbons LE, McCurry SM, Logsdon RG, Buchner DM, Barlow WE, et al. Exercise plus behavioral management in patients with Alzheimer disease: a randomized controlled trial. Jama. 2003 Oct 15;290(15):2015-22.
51. https://www.webmd.com/alzheimers/guide/alzheimers -therapies-music-art-more#1.
52. Hansen NV, Jørgensen T, Ørtenblad L. Massage and touch for dementia. Cochrane Database of Systematic Reviews. 2006(4).
53. Zafra-Tanaka JH, Pacheco-Barrios K, Tellez WA, Taype-Rondan A. Effects of dog-assisted therapy in adults with dementia: A systematic review and meta-analysis. BMC psychiatry. 2019 Dec 1;19(1):41.
54. https://www.kindlycare.com/dementia-alzheimersactivities/
55. Thompson LM. A question of balance. Nature. 2008 Apr;452(7188):707-8.
56. Mucke L. Alzheimer’s disease. Nature. 2009 Oct;461(7266):895-7.
57. Tang J, Xu H, Fan X, Li D, Rancourt D, Zhou G, et al. Embryonic stem cell-derived neural precursor cells improve memory dysfunction in Aβ (1–40) injured rats. Neuroscience Research. 2008 Oct 1;62(2):86-96.
58. Li Z, Gao C, Huang H, Sun W, Yi H, Fan X, et al. Neurotransmitter Phenotype Differentiation and Synapse Formation of Neural Precursors Engrafting in Amyloid-β 1-40 Injured Rat Hippocampus. Journal of Alzheimer’s Disease. 2010 Jan 1;21(4):1233-47.
59. Steinbeck JA, Koch P, Derouiche A, Brüstle O. Human embryonic stem cell-derived neurons establish regionspecific, long-range projections in the adult brain. Cellular and Molecular Life Sciences. 2012 Feb 1;69(3):461-70.
60. Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proceedings of the National Academy of Sciences. 2009 Aug 11;106(32):13594-9.
61. Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proceedings of the National Academy of Sciences. 2009 Aug 11;106(32):13594-9.
62. Korecka JA, Verhaagen J, Hol EM. Cell-replacement and gene-therapy strategies for Parkinson’s and Alzheimer’s disease.
63. Chakraborty A, Diwan A. Selection of Cells for Parkinson’s Disease Cell-Therapy. Int J Stem Cell Res Ther.. 2019;6:063.
64. German CL, Baladi MG, McFadden LM, Hanson GR, Fleckenstein AE. Regulation of the dopamine and vesicular monoamine transporters: pharmacological targets and implications for disease. Pharmacological Reviews. 2015 Oct 1;67(4):1005-24.